![]()
|
|
|
|
Several lines of evidence support the hypothesis that similar mechanisms are involved in the human disease. First, patients with Friedreich ataxia have iron deposits in the heart and increased mitochondrial iron. Second, fibroblasts from patients with Friedreich ataxia show enhanced sensitivity to H202. Third, a phenotype resembling Friedreich ataxia is caused by deficiency of vitamin E, a free radical scavenger localized in mitochondrial and other membranes. Fourth, oxidation-sensitive iron-sulfur cluster-containing enzymes, including respiratory complexes I, II, III, and aconitase, are defective in the heart of Friedreich ataxia patients. Fifth, MR spectroscopy analysis of skeletal muscle in patients with Friedreich ataxia showed a reduced rate of ATP synthesis after exercise, demonstrating mitochondrial dysfunction in vivo.3
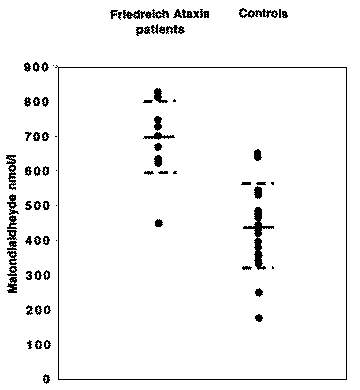
However, no study of patients with Friedreich ataxia has so far reported in vivo evidence of oxidative damage. For this purpose, we measured plasma malondialdehyde (MDA), a product of lipid peroxidation, in 11 children-three boys and eight girls-between 9.5 and 17.7 years of age (mean +/- SD 14.11 +/- 2.65) diagnosed with Friedreich ataxia according to the clinical criteria set by the Québec Collaborative Study on Friedreich ataxia.4 All were ambulatory at the time of the study. The diagnosis was confirmed by molecular testing performed by PCR.1 All patients were homozygous for expanded GAA repeats in the frataxin gene; the size of the shorter GAA repeat (GAAl) varied between 490 and 750 triplets (average 622), the size of the longer GAA repeat (GAA2) varied between 520 and 900 triplets (average 759). All patients were enrolled in a pilot, open-label trial to evaluate the toxicity and efficacy of idebenone in Friedreich ataxia. The 22 healthy controls included 8 boys and 14 girls between 6.2 and 17.4 years of age (mean +/- SD 12 +/- 0.9). The study was approved by the Ethics Committee of Ste.-Justine Hospital and consent was obtained from parents or tutors as well as from the children, after providing appropriate information for their age. Plasma MDA levels were determined by a highly sensitive and simple high-performance liquid chromatography (HPLC) method that is applicable to numerous clinical samples without any extraction procedures.5 The minimum detection level for MDA was 0.01 pmol, comparable to gas-chromatographic and fluorometric assays.5 Plasma MDA was higher in the affected children (683 +/- 105 nmol/L versus 434 +/- 26 nmol/L, p = 0.00000337 by two-tailed t-test, figure). There was no significant correlation with disease duration or with GAA expansion sizes.
|
|
Figure. Scatterplot shows plasma malondialdehyde (MDA) levels (nmol/L) in 11 patients with Friedreich ataxia versus 24 age- and sex-matched controls. In each group, the mean value is indicated with a thick line mark; +/- 1 standard devi- ation values are indicated by broken line marks. |
Whereas additional assays, like blood glutathione, glutathione peroxidase, and superoxide dismutase, which we did not perform on this series of patients, may provide a more complete picture of the antioxidant systems in Friedreich ataxia, MDA levels, by closely reflecting lipid peroxidation, may be a direct sign of active neurodegeneration involving an oxidative damage mechanism. This was clearly shown by the case of aceruloplasminemia, a disease characterized by toxic iron accumulation in the CNS. In this disease, increased plasma levels of lipid peroxidation products, mostly MDA, were detected as thiobarbituric acid reactive substances (TBARS). Treatment with desferioxamine was effective in ameliorating the neurologic deficit as well as in decreasing plasma TBARS.6 Levels of plasma MDA in patients with Friedreich ataxia may similarly provide a simple test to monitor the effect of treatments. The coenzyme Q analog idebenone has recently been proposed as treatment for Friedreich ataxia, after the observation that it can protect both membrane lipids and mitochondrial respiratory chain enzymes from iron-mediated injury in vitro.7 The report of three Friedreich ataxia patients with hypertrophic cardiomyopathy who had a markedly reduced left-ventricular mass index after 4 to 9 months of treatment with this drug bas been particularly encouraging.7 We re-tested the same 11 Children after 3 months of treatment with 5 mg/Kg/day of idebenone.7 We found no change in plasma MDA levels (from 683 +/- 105 nmol/L to 620 +/- 178 nmol/L, p = 0.35), although plasma MDA did decrease in a few patients. Among the six who had a mild hypertrophic cardiomyopathy, three were unchanged and three had a minor decrease in heart mass. Our results suggest that this dosage and length of treatment with idebenone, although sufficient to induce cardiac improvement, may be only marginally effective to reduce lipid peroxydation in vivo. Higher dosages and longer treatment may be required to reduce lipid peroxidation, which may be particularly important to affect the neurodegenerative process. Other antioxidants should also be evaluated for such activity.
From the Service de Neurologie (M. Emond and Dr. Vanasse) and the Service et Unité de Recherche en Gastroenterologie-Nutrition (Dr. Lepage), Hôpital Sainte Justine, Montréal; and the Centre Hospitalier de l'Université de Montréal (Dr Pandolfo), Québec, Canada. Supported by grants from the National Institute of Neurological Diseases and Stroke (NINDS), by the Medical Research Council of Canada (MRC), by the Muscular Dystrophy Association (MDA), USA, and by the Association Canadienne de l'Ataxie de Friedreich (ACAF). M.P. is supported by an MRC Scientist Award.
Received July 18, 2000. Accepted in final form October 12,
2000.
Address correspondence and reprint requests to Dr. Massimo Pandalfo, Centre Hospitalier de lUniversité de Montréal, Hôpital Notre-Dame-Y5608, 1560 rue Sherbrooke Est, Montréal, Québec, Canada, H2L 4M1; e-mail. massimo.pandolfo@umontreal.ca
Copyright © 2000 by AAN Enterprises,
Inc.
References
1.Campuzano V, Montermini L, Molto MD, et al. Friedreich ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996;271:1423-1427.
2. Babcock M, de Silva D, Oaks R, et al. ReguIation of mitochondriaI iron accumulation by Yfh1, a putative homolog of frataxin. Science 1997;276: 1709 1712.
3. Lodi R, Cooper JM, Bradley JL, et al. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc Natl Acad Sci USA 1999;96:11492-11495.
4. Geoffroy G, Barbeau A, Breton G, et al. Clinical description and roentgenologic evaluation of patients with Friedreich ataxia. Can J Neurol Sci 1976;3:279-286.
5. Fukunaga K, Takama K, Suzuki T. High-Performance liquid chromatographic determination of plasma malondialdehyde level without a solvent extraction procedure. Anal Biochem 1995;230:20-23.
6. Miyajima H, Takahashi Y, Kamata T, et al. Use of desferrioxamine in the treatment of aceruloplasminemia. Ann Neurol 1997;41:404-407.
7. Rustin P, von Kleist-Retzow JC, Chantrel-Groussard K, et al. Effect of idebenone on cardiomyopathy in Friedreich's ataxia: a preliminary study. Lancet 1999;354:477-479.